
Aparna Rao1, Lisa A. Schimmenti2, Elizabeth Vestal1, Cheri Schnooveld3, Melissa Ferrello3, Jennifer Ward3 and Barbara Friedman3
1 Department of Speech-Language-Hearing Sciences
164 Pillsbury Drive SE
University of Minnesota, Minneapolis, MN 55455
2 Department of Pediatrics and Ophthalmology
Division of Genetics and Metabolism
420 Delaware St. SE
University of Minnesota, Minneapolis, MN 55455
3 University of Minnesota Medical Center, Fairview
Audiology Clinic
516 Delaware St SE
Minneapolis, MN 55455
Acknowledgments
We are indebted to Alyssa Anderson for helping with consents and data management. We are also grateful for financial support received from the Lion's 5M Hearing Foundation.
Abstract
Molecular genetic testing for hearing loss is now part of the standard protocol for etiologic diagnosis of hearing loss. To be effective collaborators in the early identification process, audiologists need to be equipped with adequate information about the steps involved in etiologic diagnosis of hearing loss. Audiologists are not only involved in characterizing the hearing loss, but also in providing counseling and appropriate referrals to families. In this article, the challenge of genetic testing and the decision-making process in genetic testing are discussed. Case studies are used to highlight the clinical process for genetic testing to diagnose the etiology of hearing loss.
Introduction
Hearing loss is the most common sensory impairment in newborns, found in 1-3 per 1000 births (Mehl & Thompson, 2002; Morton & Nance, 2006). Universal newborn hearing screening has made early identification of hearing loss a reality (Harrison, Roush, & Wallace, 2003; Sininger et al., 2009). Hearing loss can result from genetic and/ or environmental factors. At least in developed nations, improved medical care during pregnancy and early life has led to a reduction in the prevalence of hearing loss due to environmental factors. Therefore, a majority of cases with hearing loss in early life is due to a genetic etiology (Morton & Nance, 2006; Toriello, Reardon, & Gorlin, 2004). Both the Joint Committee on Infant Hearing (JCIH, 2007) and the American College of Medical Genetics (ACMG, 2002) recommend genetic testing for infants identified with hearing loss.
An important benefit of genetic testing is etiologic diagnosis of hearing loss (JCIH, 2007; ACMG, 2002). Both families and physicians gain invaluable information that can guide medical management and intervention of babies with hearing loss (Robin, Prucka, Woolley, & Smith, 2005; Withrow et al., 2009). A frequently reported motivation of families seeking genetic testing is the understanding of the cause of the hearing loss (Withrow et al., 2009). Identification of the cause can direct medical management; for example, in cases with syndromic hearing loss, medical monitoring of associated health concerns is paramount. An etiology can also help parents make decisions regarding interventions such as cochlear implants. In addition, the psychological benefit of knowing the cause of the hearing loss cannot be underestimated for families. An associated benefit of genetic evaluation and testing for families includes provision of timely information regarding the probability of recurrence in future offspring and improving early diagnosis for potentially affected siblings. Establishing the etiology of hearing loss through genetic testing can also eliminate further expenditure in diagnostic healthcare costs (Preciado et al., 2005). Today, genetic evaluation and testing for hearing loss is an established step in the etiologic diagnosis of hearing loss (JCIH, 2007). This article is directed to those with introductory knowledge in genetics. A list of websites in Appendix A may be helpful to those who intend to gain basic information in genetics.
The Challenge of Genetic Testing
Owing partly to the human genome project, over 110 chromosomal loci and more than 65 genes have been identified to cause hearing loss in the last two decades (Van Camp & Smith, 2011). Therefore, hearing loss exhibits locus heterogeneity as it is not a single genetic entity, but can result from different genetic causes. The term "deafness associated genetic variants" may be used to describe mutations, especially when counseling families, as the word mutation may carry a negative connotation for some. Current technology for clinical testing allows for testing only one gene or a few genes in a sequential order. DNA sequence analysis used to characterize the nucleotide sequence of a gene is recommended to detect all known mutations in the gene and to detect deletions (del Castillo et al., 2002) that may occur in some cases. As hearing loss may be caused by multiple mutations in multiple genes, the test(s) to be chosen presents a challenge for the geneticist. Moreover, the cost associated with genetic testing contributes to the responsibility to order the most appropriate test. Analysis of several genes in a single test may be achieved by using next-generation sequencing where many genes can be sequenced at the same time. This exciting technology is currently limited to research use but we anticipate its clinical availability presenting in the not so distant future (Shearer et al., 2010).
Genetics of Hearing Loss
Hearing loss may be classified based on presence or absence of associated medical conditions (syndromic vs. nonsyndromic) and mode of inheritance (dominant, recessive, x-linked, and mitochondrial). In approximately 30% of the cases, the hearing loss is syndromic, occurring with other medical conditions (Toriello, Reardon, & Gorlin, 2004). Nonsyndromic hearing loss, wherein hearing loss is the sole symptom, accounts for approximately 70% of the cases. The inheritance pattern of hearing loss in a majority of cases is autosomal recessive (77%); autosomal dominant (22%), x-linked (1%) and mitochondrial mutations (
More than 400 syndromes have been identified with hearing loss as one of the symptoms (Toriello, Reardon & Gorlin, 2004). There are several sources that offer a comprehensive review of syndromes; they are listed in Appendix A. Features of syndromes relevant to the current discussion are summarized in Table 1.
At this time, clinical genetic testing is not available for all the genes listed. The list of genetic tests for etiologic diagnosis of hearing loss keeps growing rapidly; the reader is encouraged to visit the website www.genetests.org/ for current information regarding genetic tests and laboratories that perform these tests on a clinical or research basis. 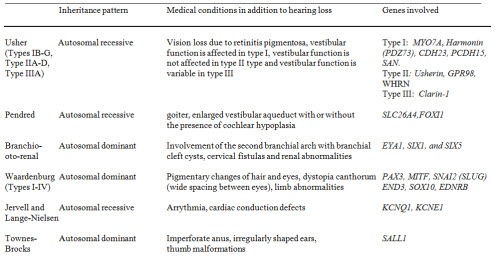
Table 1. Summary of syndromes associated with hearing loss described in this review. Click Here to View a Larger Version of Table 1 (PDF)
Kelsell et al. (1997) discovered the first gene for nonsyndromic hearing loss. This gene, known as Gap Junction Beta 2 (GJB2), is typically transmitted in an autosomal recessive fashion, although it may also be transmitted in an autosomal dominant pattern. Mutations in the gene GJB2 are known to account for 30-50% of all cases with nonsyndromic recessive hearing loss (Denoyelle et al., 1997), making it the most frequent cause of hearing loss in infants. GJB2 encodes the gap junction protein, Connexin 26, found in the supporting cells, connective tissue and other cells of the cochlea. They help form intercellular gap junction or channels between cells and may play a role in the recycling of potassium ions from the hair cells to the stria vascularis (Martin, Coleman, Casalotti, Forge & Evans, 1999). The GJB2 hearing loss phenotype is typically prelingual, bilaterally symmetrical, and severe to profound in degree (Denoyelle et al., 1999; Cryns et al., 2004). Some affected individuals may have hearing loss in the moderate to mild range (Denoyelle et al., 1999; Cryns et al., 2004; Snoeckx et al., 2005). Progression of hearing loss is observed in approximately 1/3rd of the patients (Cohn et al., 1999; Cohn & Kelley, 1999). Asymmetry and variability in the degree and configuration among siblings has also been reported (Snoeckx et al., 2005). Vestibular function is typically not affected (Denoyelle et al., 1999). More than 110 syndromic and nonsyndromic mutations of GJB2 have been reported. A list of GJB2 mutations is available at the Connexin deafness homepage (davinci.crg.es/deafness/). The mutations 35delG, 167delT, and 235delC have been shown to occur most frequently in Caucasians, Ashkenazi Jews and Asian populations respectively. The specific mutation 35delG accounts for 28% to 63% of the mutant alleles in Caucasians (Gaspirini et al., 2000).
A mutation in the gene GJB2 on one chromosome inherited along with a second mutation, usually a large deletion in the gene GJB6, located on the same chromosome (13q) will cause hearing loss. GJB6 is responsible for encoding Connexin 30, which combine to form gap junctions with Connexin 26 (Ahmad, Chen, Sun & Lin, 2003). Two large deletions in GJB6 have been found; delGJB6-D13S1830 and delGJB6-D13S1854 (del Castillo et al., 2002, 2003, 2005; Lerer et al., 2001). Recent evidence demonstrates loss of expression of GJB2 by the delGJB6-D13S1830 deletion in GJB6 (Rodriguez-Paris & Schrijver, 2009).
Today, more than 30 genes have been associated with recessive hearing loss, apart from the two main Connexin genes discussed above. Also, 24 genes associated with autosomal dominant nonsyndromic hearing loss and two genes associated with x-linked nonsyndromic hearing loss have been identified. Mitochondrial genes may also cause nonsyndromic hearing loss, particularly if an individual is exposed to aminoglycoside antibiotics (Prezant et al., 1993). A comprehensive list of genes associated with hearing loss may be found at the hereditary hearing loss homepage (hereditaryhearingloss.org/). This webpage also offers links to the function of these genes. As mentioned earlier, it is important to remember that some of the genes may show both dominant and recessive patterns of inheritance (for example, specific GBJ2 mutations may be associated with either dominant or recessive inheritance). It is also critical to note that clinical tests are not available for all genes that cause hearing loss. The website mentioned above (www.genetests.org), has updated information on clinical tests available to detect nonsyndromic hearing loss.
Genetic Testing for Hearing Loss
A team approach involving an otolaryngologist, geneticist, genetic counselor, ophthalmologist and an audiologist is recommended to diagnose the etiology in a child with hearing loss (JCIH, 2007). The well-accepted first steps toward the etiologic diagnosis of babies with congenital hearing loss are a comprehensive history, physical examination and audiological evaluation. This first stage of evaluation can help the diagnostician determine the necessity for studies to determine prenatal infections such as toxoplasmosis or cytomegalovirus. Following this basic protocol can also identify many syndromes with hearing loss as one of the features. If a syndrome can be identified based on the array of clinical symptoms, genetic testing can be limited to the relevant set of mutations that are known to cause a syndrome. For instance, when dystopia canthorum (wide spacing between the eyes) and pigmentary changes in the hair and eyes are present with hearing loss, testing for Waardenburg syndrome would be considered. Case history of delayed walking in a child with profound hearing loss would direct the diagnostician to test for Usher syndrome type I, subtype B. However, the problem of choosing the appropriate genetic test remains if the associated clinical features, with the exception of hearing loss, are not present in early life. In such instances, the diagnostician has to rely on medical diagnostic tests to improve the yield of genetic testing. The most frequently used diagnostic tests for sensorineural hearing loss (SNHL) are a CT scan of the cochlear and vestibular structures, renal ultrasound, electrocardiogram, thyroid studies, and ophthalmological testing.
An enlarged vestibular aqueduct (EVA) is the most common inner ear deformity associated with SNHL that can be visualized by CT scan of the temporal bone. EVA may occur in isolation or in combination with other malformations of the inner ear (Atkin, Grimmer, Hedlund, & Park, 2009). When the CT scan is suggestive of EVA with or without Mondini deformity the recommendation is to screen for SLC26A4. Mutations in the SLC26A4 gene have been found in nearly 40% of the individuals with EVA (Albert et al., 2006). Biallelic mutations (i.e., mutations in both alleles) were found in 24% of the subjects studied, and monoallelic mutations were found in 16% of the subjects studied. Not all cases with the SLC26A4 mutation will develop goiter associated with Pendred syndrome. However, a positive test for SLC26A4 will trigger the need to regular monitoring for thyroid function (Choi et al., 2011).
In the presence of clinical features such as pinnae malformations or branchial arch remnants, a renal ultrasound may also be performed. For example, in cases with branchio-oto-renal (BOR) syndrome, in addition to malformations of the external and middle ear, renal abnormalities may be present. Diagnosis of the renal condition through renal ultrasound will help a geneticist choose the appropriate genetic test with greater confidence (Chang et al., 2004).
An EKG is frequently conducted in deaf children; however, sometimes a cardiac evaluation is completed only if there is a family history of syncope. Another frequently reported strategy is to perform an EKG when test results for other possible genetic conditions are negative. The goal is to diagnose Jervell and Lange-Nielsen syndrome which presents with cardiac defects (prolonged QT interval) and hearing loss. With timely diagnosis, medical management for the cardiac issues can begin and prevent sudden death, which in most cases is the first sign of this condition (Schwartz et al., 2006).
In a majority of cases with SNHL, the family history does not reveal other affected family members and there are no associated clinical features. When a child presents with hearing loss of unknown etiology that appears to be nonsyndromic and recessive, testing for mutations in the GJB2 gene by sequence analysis, based on the frequency of occurrence of the mutation is recommended (ACMG, 2002; McGuirt & Smith, 1999; Kenna et al., 2007). The ethnicity of the patient is an important variable. Specific mutations are known to be prevalent in certain populations (for example, the 35delG mutation is predominant in the Caucasian population). While it is useful to screen for common mutations relevant to the target population, it is advised that all mutations be tested if the first screen is negative or if the patient is heterozygous (i.e., has two different forms of the same gene). Today, most laboratories perform sequence analysis of a gene that is able to detect all possible mutations. Since this is a recessive condition, if two mutations known to be pathogenic are found in a gene, then the etiology of the hearing loss is certain. In cases that are nonsyndromic and recessive, the most frequent genetic causes are due to mutations in GJB2 and GJB6.
Although Connexin 26 testing is routinely recommended in the absence of ear malformations and other disorders, Kenna and colleagues (2007) have found other conditions such as neurocognitive disorders, urologic abnormalities, structural malformations of the external and/ or the middle ear, and subtle abnormalities of the inner ear in children with mutations of GJB2. The authors caution that the size of the cohort in this study was small and these may be unrelated etiologies, but recommend testing for GJB2 mutations in the presence of these disorders.
Candidate genes for genetic testing may be selected based on the phenotype. The diagnosis of auditory neuropathy/dys-synchrony points the geneticist to screen for mutations in the genes otoferlin (OTOF) (Varga et al., 2003; Rodríguez-Ballesteros et al., 2008) and Pejvakin (Delmaghani et al., 2006). Both genes are transmitted in an autosomal recessive pattern. When low-frequency hearing loss is diagnosed and autosomal dominant transmission is seen, screening for mutations in the gene WFS1 is indicated. When genetic testing results for known genes are negative, a recommended strategy is to rule out Usher syndrome so that treatment can begin, if the condition is diagnosed (Hilgert, Smith, & Van Camp, 2009). Genetic testing for early diagnosis of Jervell and Lange-Nielsen syndrome should also be considered, especially if the EKG test results are borderline normal (Wang, Bowles & Towbin, 1998).
In summary, genetic testing may be guided by the case history, phenotype, and the relative prevalence of a gene in a clinical population. Results of medical tests may help improve the yield of clinical genetic testing.
Case Studies to Highlight Clinical Decision Making
Case studies have been chosen to highlight the process of decision making for genetic testing. A team of professionals including audiologists, otorhinolaryngologists, infectious disease specialist, geneticist, genetic counselor, speech-language pathologist and developmental pediatrician evaluated these cases. All families provided informed consent to be included in a database set up to study hearing loss in children. Approval was also received from the Institutional Review Board at the University of Minnesota.
Case one.
Baby A failed the newborn hearing screening in both ears before hospital discharge. However, four weeks later, he passed the rescreening in both ears. At 1 year of age, parental concern triggered a referral for a comprehensive audiological evaluation. At this time, the child was diagnosed with severe-to-profound hearing loss in both ears and was fit with amplification soon after diagnosis. A CT scan conducted as part of etiologic diagnosis of hearing loss revealed normal temporal bone anatomy in both ears. An EKG was found to be within normal limits. Syndromic features were absent. Family history obtained by the genetic counselor revealed no other affected family members and the ethnicity was Caucasian. The baby was tested for mutations in the gene GJB2 and was found to be homozygous (i.e., has identical forms of the same gene) for the mutation 35delG. The genetic counselor presented the results to the parents.
The child received the first cochlear implant after a 3-month hearing aid trial and the second implant 18 months later. Intervention is being provided in an auditory-verbal setting with documented gains in speech and language skills.
The decision for genetic testing in this case is uncomplicated. The autosomal recessive pattern and a normal CT prompted testing for mutations in GJB2. As the child was found to be homozygous for mutation 35delG, it is confirmatory as the cause for the hearing loss. As mutations in GJB2 cause non-syndromic hearing loss, this child is not at risk for other health concerns that are associated with sensorineural hearing loss. The estimated risk for future offspring in one is four. Children with GJB2 mutations are known to perform well with cochlear implants (Wrobel et al., 2008; Sinnathuray et al., 2004a, b).
Case two.
Child B was first seen at 2 years of age. He presented a history of mild to moderate hearing loss, which was diagnosed soon after birth. Consistent hearing aid use was recorded at 11 months of age and he received auditory-oral intervention. Previous history revealed that genetic testing was done for GJB2 mutations and was found to be negative.
A CT scan obtained when the child was 2 years of age revealed EVA bilaterally. His EKG was found to be normal. Progression was noted in his hearing loss; it was now moderate to severe in the right ear and moderately severe to profound in the left ear. Family history review revealed no other affected family members. Based on the result on the CT scan and the family history, genetic testing was conducted for SLC26A4. Two pathologic mutations were found in the SLC26A4 gene. Based on the results of the genetic test, thyroxin and thyroid-stimulating hormone were checked and found to be normal.
Recommendations include annual thyroid testing and observation for thyroid enlargement. Hearing aids were re-programmed to his hearing thresholds; however, parents are considering a cochlear implant for the right ear. He is currently receiving intervention in an auditory-oral setting with progress being reported in speech and language skills.
Based on the phenotype presented (presence of EVA), testing was conducted for SLC26A4. Recommendations were made based on the positive results for mutations in SLC26A4 that includes monitoring of thyroid function.
Case three.
Sibling A was first seen at 11 years of age for a cochlear implant evaluation. She was diagnosed with severe to profound hearing loss in both ears and was fit with amplification. She was using a combination of oral and cued speech and the amplification was not adequate for her listening needs. When she came in for a cochlear implant evaluation, she presented a history of childhood hearing loss in the family. Her sister (sibling B), aged 4-months of age, was diagnosed with severe-profound hearing loss as well.
A thorough case history revealed an autosomal recessive pattern and the ethnicity was Caucasian. Sibling A's CT scan results were equivocal for EVA. Sibling B was diagnosed with mild EVAs bilaterally.
Based on the results of the CT scan, sibling A was first tested for SLC26A4; the result was found to be negative. Genetic testing was then done for GJB2 mutations and was found to be homozygous for 35delG. After this result was received, sibling B was also tested and found to be homozygous for 35delG.
The likelihood of a genetic etiology is high when both children in the family have severe-profound hearing loss. Testing for genetic hearing loss can be conducted at any age, and in this case, the older sibling was 11 years old with a history of congenital hearing loss. The decision to test for SLC26A4 in the older sibling was based on the possibility of EVA. However, when the genetic test for SLC26A4 was negative, testing for mutations in GJB2 was the logical next step due to the relatively high prevalence of this genetic condition. The most commonly occurring mutation in the Caucasian population is 35delG. Both sisters were homozygous for 35delG making it certain that this genetic variation had caused the hearing loss.
Case four.
Case D was seen for evaluation of right unilateral atresia and microtia. Hearing was found to be within normal limits in the left ear. Masked bone-conduction thresholds in the right ear were within normal limits as well. Both, the atresia and microtia were classified as grade III. A CT scan of the right ear revealed deformation and fusion of the malleus and incus. The right vestibule was found to be bulbous and mildly dilated. This child did not have other medical conditions. Genetic testing was conducted for EYA1 (BOR syndrome) and SALL1 (Townes-Brocks syndrome) based the physical findings. Both tests were found to be negative. A negative result on a genetic test does not preclude the possibility that the hearing loss has a genetic origin. Parents should be encouraged to revisit the possibility in the future; advancements in knowledge about genetic hearing loss and test techniques may provide more options to determine the etiology of the hearing loss.
Summary and Caveats Related to Genetic Testing
Advances in genetic testing have transformed our diagnostic approach to hearing loss in infants. Genetic testing is a complex process; it takes knowledge and experience to obtain the information needed in a time effective and cost effective manner. The clinical geneticist and genetic counselor are indispensable to the process. There are certain caveats related to genetic testing that audiologists need to be aware of. Positive test results are typically highly accurate, although ambiguities may exist in the interpretation of newly recognized mutations. Negative results may not always rule out the diagnosis of a disorder or a genetic cause for hearing loss. Mutations in segments of DNA that do not make up a gene (non-coding regions) cannot be ruled out as a potential cause or risk factor as they may be involved in regulating genes that cause hearing loss. Lastly, it is important to remember that families are best served when genetic testing and results are presented in a genetic counseling setting by a qualified geneticist and genetic counselor. The main reason for genetic testing is to provide medically relevant information for the care of a child who deaf or hard of hearing. In the examples above, genetic testing provided an answer for families as to why their child has hearing loss and provided a framework for medical care.
Appendix A
Websites recommended for an introduction to genetics:
genome.gov/
www.nchpeg.org/shla/site.html
Websites recommended to learn about syndromes:
www.nidcd.nih.gov/health/hearing/
ghr.nlm.nih.gov/
References
American College of Medical Genetics. (2002). Genetics evaluation guidelines for the etiologic diagnosis of congenital hearing loss. Genetic Evaluation of Congenital Hearing Loss Expert Panel. ACMG statement. Genetics in Medicine, 4, 162-171.
Albert, S., Blons, H., Jonard, L., Feldmann, D., Chauvin, P., Loundon, N., et al. (2006). SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. European Journal of Human Genetics, 14, 773-779.
Ahmad, S., Chen, S., Sun, J., & Lin, X. (2003). Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochemical and Biophysical Research Communications, 307, 362-368.
Atkin, J.S., Grimmer, J.F., Hedlund, G., & Park, A.H. (2009). Cochlear abnormalities associated with enlarged vestibular aqueduct anomaly. International Journal of Pediatric Otorhinolaryngology, 73, 1682-1685.
Chang, E. H., Menezes, M., Meyer, N. C., Cucci, R. A., Vervoort, V. S., Schwartz, C. E., et al. (2004). Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Human Mutation, 23, 582-589.
Choi, B. Y., Muskett, J., King, K. A., Zalewski, C. K., Shawker, T., Reynolds, J. C., et al. (2011). Hereditary hearing loss with thyroid abnormalities. Advances in Otorhinolaryngology, 70, 43-49.
Cohn, E.S., & Kelley, P.M. (1999). Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most common cause of childhood hearing loss. American Journal of Medical Genetics, 89, 130-136.
Cohn, E.S., Kelley, P.M., Fowler, T.W., Gorga, M.P., Lefkowitz, D.M., Kuehn, H.J., et al. (1999). Clinical studies of families with hearing loss attributable to mutations in the connexin 26 gene (GJB2/DFNB1). Pediatrics, 103, 546-550.
Cryns, K., Orzan, E., Murgia, A., Huygen, P. L., Moreno, F., del Castillo, I., et al. (2004). A genotype-phenotype correlation for GJB2 (connexin 26) deafness. Journal of Medical Genetics, 41, 147-154.
del Castillo, F.J., Rodriguez-Ballesteros, M., Alvarez, A., Hutchin, T., Leonardi, E., de Oliveira, C.A., et al. (2005) A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. Journal of Medical Genetics, 42, 588-594.
del Castillo, I., Moreno-Pelayo, M.A., del Castillo, F.J., Brownstein, Z., Marlin, S., Adina, Q. et al. (2003). Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. American Journal of Human Genetics, 73, 1452-1458.
del Castillo, I., Villamar, M., Moreno-Pelayo, M.A., del Castillo, F.J., Alvarez, A., Telleria, D., et al. (2002) A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. New England Journal of Medicine, 346, 243-249.
Delmaghani, S., del Castillo, F. J., Michel, V., Leibovici, M., Aghaie, A., Ron, U. et al.(2006). Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nature Genetics, 38, 770-778.
Denoyelle, F., Marlin, S., Weil, D., Moatti, L., Chauvin, P., Garabedian, E. N., et al. (1999). Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: implications for genetic counselling. Lancet, 353, 1298-1303.
Denoyelle, F, Weil, D, Maw, M.A., Wilcox, S.A., Lench, N.J., Allen-Powell, D.R. et al. (1997). Prelingual deafness: High prevalence of a 30delG mutation in the connexin 26 gene. Human Molecular Genetics, 6, 2173-2177.
Gasparini, P., Rabionet, R., Barbujani, G., Melchionda, S., Petersen, M., Brondum-Nielsen, K., et al. (2000). High carrier frequency of the 35delG deafness mutation in European populations. European Journal of Human Genetics, 8, 19-23.
Harrison, M., Roush, J., & Wallace, J. (2003). Trends in age of identification and intervention in infants with hearing loss. Ear and Hearing, 24, 89-95.
Hilgert, N., Smith, R. J., & Van Camp, G. (2009). Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutation Research, 681, 189-196.
Joint Committee on Infant Hearing. (2007). Year 2007 position statement: Principles and guidelines for early hearing detection and intervention programs. Pediatrics, 120, 898-921.
Kelsell, D.P., Dunlop, J., Stevens, H.P., Lench, N.J., Liang, J.N., Parry, G., et al. (1997). Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature, 6628, 80-83.
Kenna, M.A., Rehm, H.L., Robson, C.D., Frangulov, A., McCallum, J., Yaeger, D., et al. (2007). Additional clinical manifestations in children with sensorineural hearing loss and biallelic GJB2 mutations: who should be offered GJB2 testing? American Journal of Medical Genetics, 143A, 1560-1566.
Lerer, I., Sagi, M., Ben-Neriah, Z., Wang, T., Levi, H., Abeliovich, D. (2001). A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: A novel founder mutation in Ashkenazi Jews. Human Mutation, 18, 460.
Martin, P. E., Coleman, S. L., Casalotti, S. O., Forge, A., & Evans, W. H. (1999). Properties of connexin26 gap junctional proteins derived from mutations associated with non-syndromal hereditary deafness. Human Molecular Genetics, 8, 2369-2376
McGuirt, W.T., & Smith, R.J. (1999). Connexin 26 as a cause of hereditary hearing loss. American Journal of Audiology, 8, 93-100.
Mehl, A.L., & Thomson, V. (2002). The Colorado newborn hearing screening project, 1992-1999: on the threshold of effective population-based universal newborn hearing screening. Pediatrics, 109, E7.
Morton, C.C. (2002). Genetics, genomics and gene discovery in the auditory system. Human Molecular Genetics, 11, 1229-1240.
Morton, C.C., & Nance, W.E. (2006). Newborn hearing screening--a silent revolution. New England Journal of Medicine, 354, 2151-2164.
Prezant, T. R., Agapian, J. V., Bohlman, M. C., Bu, X., Oztas, S., Qiu, W. Q., et al. (1993). Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nature Genetics, 4, 289-294.
Preciado, D.A., Lawson, L., Madden, C., Myer, D., Ngo, C., Bradshaw, J.K., et al. (2005). Improved diagnostic effectiveness with a sequential diagnostic paradigm in idiopathic pediatric sensorineural hearing loss. Otology and Neurotology, 26, 610-615.
Robin, N.H., Prucka, S.K., Woolley, A.L., & Smith, R.J. (2005). The use of genetic testing in the evaluation of hearing impairment in a child. Current Opinion in Pediatrics, 17, 709-712.
Rodriguez-Ballesteros, M., Reynoso, R., Olarte, M., Villamar, M., Morera, C., Santarelli, R. et al. (2008). A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Human Mutation, 29, 823-831.
Rodriguez-Paris, J., & Schrijver, I. (2009). The digenic hypothesis unraveled: the GJB6 del(GJB6-D13S1830) mutation causes allele-specific loss of GJB2 expression in cis. Biochemical and Biophysical Research Communications, 389, 354-359.
Shearer, A. E., DeLuca, A. P., Hildebrand, M. S., Taylor, K. R., Gurrola, J., 2nd, Scherer, S., et al. (2010). Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proceedings of the National Academy of Sciences, 107, 21104-21109.
Schwartz, P. J., Spazzolini, C., Crotti, L., Bathen, J., Amlie, J. P., Timothy, K., et al. (2006). The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation, 113, 783-790.
Sininger, Y.S., Martinez, A., Eisenberg, L., Christensen, E., Grimes, A., & Hu, J. (2009). Newborn hearing screening speeds diagnosis and access to intervention by 20-25 months. Journal of the American Academy of Audiology, 20, 49-57.
Sinnathuray, A.R., Toner, J.G., Clarke-Lyttle, J., Geddis, A., Patterson, C.C., & Hughes, A.E. (2004a). Connexin 26 (GJB2) gene-related deafness and speech intelligibility after cochlear implantation. Otology and Neurotology, 25, 935-942.
Sinnathuray, A.R., Toner, J.G., Geddis, A., Clarke-Lyttle, J., Patterson, C.C., & Hughes, A. E. (2004b). Auditory perception and speech discrimination after cochlear implantation in patients with connexin 26 (GJB2) gene-related deafness. Otology and Neurotology, 25, 930-934.
Snoeckx, R.L., Huygen, P.L., Feldmann, D., Marlin, S., Denoyelle, F., Waligora, J., et al. (2005). GJB2 mutations and degree of hearing loss: a multicenter study. American Journal of Human Genetics, 77, 945-957.
Toriello, H.V., Reardon, W., & Gorlin, R.J. (2004). Hereditary hearing loss and its syndromes 2nd ed. New York: Oxford University Press.
Van Camp, G. & Smith, R.J.H. (2011). Hereditary Hearing Loss Homepage. Retrieved April 10, 2011 from: hereditaryhearingloss.org
Varga, R., Kelley, P. M., Keats, B. J., Starr, A., Leal, S. M., Cohn, E., et al. (2003). Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. Journal of Medical Genetics, 40, 45-50.
Wang, Q., Bowles, N. E., & Towbin, J. A. (1998). The molecular basis of long QT syndrome and prospects for therapy. Molecular Medicine Today, 4, 382-388.
Withrow, K.A., Tracy, K.A., Burton, S.K., Norris, V.W., Maes, H.H., Arnos, K.S., et al. (2009). Impact of genetic advances and testing for hearing loss: results from a national consumer survey. American Journal of Medical Genetics, 149A, 1159-1168.
Wrobel, M., Magierska-Krzyszton, M., Szyfter, K., Mietkiewska, D., Szyfter, W., Rydzanicz, M., & Karlik, M. (2008). Comparison of rehabilitation results in deaf patients with and without genetically related hearing loss. Cochlear Implants International, 9, 132-142.
Genetic Testing in Childhood Hearing Loss: Review and Case Studies
July 18, 2011
Continued and its subsidiaries provide professional education authored by qualified Subject Matter Experts for continuing education purposes. These materials are intended for educational purposes and do not constitute medical advice or a substitute for individual clinical judgment. Continued is not a clinical healthcare provider; the licensed professional is solely responsible for ensuring that the application of any techniques or information presented is within their legal scope of practice and jurisdictional requirements.

Related Courses
1
https://www.audiologyonline.com/audiology-ceus/course/grand-rounds-pediatric-phoenix-40715
Case Studies in Pediatric Audiology: Wins Worth Sharing, in partnership with Phoenix Children’s Hospital
This grand rounds presentation shares case studies of five pediatric patients seen in the Audiology Clinic at Phoenix Children’s. Evidenced based practice, family centered care, patience, and innovation were used to obtain positive patient outcomes.
auditory, textual, visual
129
USD
Subscription
Unlimited COURSE Access for $129/year
OnlineOnly
AudiologyOnline
www.audiologyonline.com
Case Studies in Pediatric Audiology: Wins Worth Sharing, in partnership with Phoenix Children’s Hospital
This grand rounds presentation shares case studies of five pediatric patients seen in the Audiology Clinic at Phoenix Children’s. Evidenced based practice, family centered care, patience, and innovation were used to obtain positive patient outcomes.
40715
Online
PT90M
Case Studies in Pediatric Audiology: Wins Worth Sharing, in partnership with Phoenix Children’s Hospital
Presented by Tanner Robinson, AuD, CCC-A, Robert Fanning, AuD, CCC-A, Alissa Nickerson, AuD, CCC-A, Ashley Geske, AuD, CCC-A, Allie Sayer, AuD, CCC-A, Wendy Steuerwald, AuD, CCC-A
Course: #40715Level: Advanced1.5 Hours
AAA/0.15 Advanced; ACAud inc HAASA/1.5; AG Bell - LSLS/1.5 Domain 1, Domain 2, Domain 5; AHIP/1.5; BAA/1.5; CAA/1.5; Calif. SLPAB/1.5; IACET/0.2; IHS/1.5; Kansas, LTS-S0035/1.5; NZAS/2.0; SAC/1.5; TX TDLR, #142/1.5 Non-manufacturer
This grand rounds presentation shares case studies of five pediatric patients seen in the Audiology Clinic at Phoenix Children’s. Evidenced based practice, family centered care, patience, and innovation were used to obtain positive patient outcomes.
2
https://www.audiologyonline.com/audiology-ceus/course/grand-rounds-phoenix-childrens-39703
Pediatric Grand Rounds: Embracing the Unexpected, in partnership with Phoenix Children’s Hospital
This pediatric grand rounds presentation presents case studies of six patients with hearing concerns. Collaboration, evidence-based practice, and innovation are used to obtain the best outcomes.
auditory, textual, visual
129
USD
Subscription
Unlimited COURSE Access for $129/year
OnlineOnly
AudiologyOnline
www.audiologyonline.com
Pediatric Grand Rounds: Embracing the Unexpected, in partnership with Phoenix Children’s Hospital
This pediatric grand rounds presentation presents case studies of six patients with hearing concerns. Collaboration, evidence-based practice, and innovation are used to obtain the best outcomes.
39703
Online
PT90M
Pediatric Grand Rounds: Embracing the Unexpected, in partnership with Phoenix Children’s Hospital
Presented by Deborah Flynn, AuD, CCC-A, Allie Sayer, AuD, CCC-A, Christina Dubas, AuD, CCC-A, Rachel Worcester, AuD, ABA-C, Caroline Sabatino, AuD, CCC-A, Robert Fanning, AuD, CCC-A, Wendy Steuerwald, AuD, CCC-A
Course: #39703Level: Advanced1.5 Hours
AAA/0.15 Advanced; ACAud inc HAASA/1.5; AG Bell - LSLS/1.5 Domain 1, Domain 2; BAA/1.5; CAA/1.5; Calif. SLPAB/1.5; IACET/0.2; IHS/1.5; Kansas, LTS-S0035/1.5; NZAS/2.0; SAC/1.5
This pediatric grand rounds presentation presents case studies of six patients with hearing concerns. Collaboration, evidence-based practice, and innovation are used to obtain the best outcomes.
3
https://www.audiologyonline.com/audiology-ceus/course/grand-rounds-nationwide-childrens-39706
Pediatric Grand Rounds: Beyond the Basics to Maximize Outcomes, presented in partnership with Nationwide Children’s Hospital
This Grand Rounds session features audiologists and speech pathologists who specialize in diagnosing and managing pediatric patients. During this course, a panel of speech and audiology professionals shares 5 clinically applicable cases that helped them grow and innovate their clinical practice. The importance of evidence-based care is highlighted to achieve the best outcomes for pediatric patients.
auditory, textual, visual
129
USD
Subscription
Unlimited COURSE Access for $129/year
OnlineOnly
AudiologyOnline
www.audiologyonline.com
Pediatric Grand Rounds: Beyond the Basics to Maximize Outcomes, presented in partnership with Nationwide Children’s Hospital
This Grand Rounds session features audiologists and speech pathologists who specialize in diagnosing and managing pediatric patients. During this course, a panel of speech and audiology professionals shares 5 clinically applicable cases that helped them grow and innovate their clinical practice. The importance of evidence-based care is highlighted to achieve the best outcomes for pediatric patients.
39706
Online
PT90M
Pediatric Grand Rounds: Beyond the Basics to Maximize Outcomes, presented in partnership with Nationwide Children’s Hospital
Presented by Gina M. Hounam, PhD, Holly T. Gerth, AuD, Lauren Durinka, AuD, Christine Schafer, AuD, Alecia Jayne, AuD, CCC-A, Ursula M. Findlen, PhD, Caitlin Cummings, PhD, CCC-SLP, Lauren Y. Yoshihiro, MS, CCC-SLP
Course: #39706Level: Advanced1.5 Hours
AAA/0.15 Advanced; ACAud inc HAASA/1.5; AG Bell - LSLS/1.5 Domain 1, Domain 2; BAA/1.5; CAA/1.5; Calif. SLPAB/1.5; IACET/0.2; IHS/1.5; Kansas, LTS-S0035/1.5; NZAS/2.0; SAC/1.5
This Grand Rounds session features audiologists and speech pathologists who specialize in diagnosing and managing pediatric patients. During this course, a panel of speech and audiology professionals shares 5 clinically applicable cases that helped them grow and innovate their clinical practice. The importance of evidence-based care is highlighted to achieve the best outcomes for pediatric patients.
4
https://www.audiologyonline.com/audiology-ceus/course/educational-management-unilateral-hearing-loss-39985
From Clinic to Classroom: Functional Approaches to Working with Children with Unilateral Hearing Loss Pt. 2, in partnership with Educational Audiology Association
This is a two-part course focusing on the diagnosis and management of pediatric unilateral hearing loss in the clinical setting, the impact of unilateral hearing loss in the educational setting, and building professional working relationships to best support our patients/students. This course reviews the medical and educational impact of unilateral hearing loss for children, current recommendations regarding device and rehabilitation options, and conducting functional testing in the clinic and school settings to provide individualized recommendations for families and educational teams.
auditory, textual, visual
129
USD
Subscription
Unlimited COURSE Access for $129/year
OnlineOnly
AudiologyOnline
www.audiologyonline.com
From Clinic to Classroom: Functional Approaches to Working with Children with Unilateral Hearing Loss Pt. 2, in partnership with Educational Audiology Association
This is a two-part course focusing on the diagnosis and management of pediatric unilateral hearing loss in the clinical setting, the impact of unilateral hearing loss in the educational setting, and building professional working relationships to best support our patients/students. This course reviews the medical and educational impact of unilateral hearing loss for children, current recommendations regarding device and rehabilitation options, and conducting functional testing in the clinic and school settings to provide individualized recommendations for families and educational teams.
39985
Online
PT60M
From Clinic to Classroom: Functional Approaches to Working with Children with Unilateral Hearing Loss Pt. 2, in partnership with Educational Audiology Association
Presented by Kate Jablonski, AuD
Course: #39985Level: Introductory1 Hour
AAA/0.1 Introductory; ACAud inc HAASA/1.0; AG Bell - LSLS/1.0 Domain 1, Domain 2, Domain 8; BAA/1.0; CAA/1.0; Calif. SLPAB/1.0; IACET/0.1; IHS/1.0; Kansas, LTS-S0035/1.0; NZAS/1.0; SAC/1.0
This is a two-part course focusing on the diagnosis and management of pediatric unilateral hearing loss in the clinical setting, the impact of unilateral hearing loss in the educational setting, and building professional working relationships to best support our patients/students. This course reviews the medical and educational impact of unilateral hearing loss for children, current recommendations regarding device and rehabilitation options, and conducting functional testing in the clinic and school settings to provide individualized recommendations for families and educational teams.
5
https://www.audiologyonline.com/audiology-ceus/course/hearing-loss-in-ccmv-38714
Hearing Loss in cCMV, in partnership with Midwestern University and Phoenix Children's Hospital
Hearing loss in cCMV has unique features, presentation, and long-term outcomes. This presentation highlights these unique features, and outlines the current evidence that guides identification and treatment of patients with this condition.
auditory, textual, visual
129
USD
Subscription
Unlimited COURSE Access for $129/year
OnlineOnly
AudiologyOnline
www.audiologyonline.com
Hearing Loss in cCMV, in partnership with Midwestern University and Phoenix Children's Hospital
Hearing loss in cCMV has unique features, presentation, and long-term outcomes. This presentation highlights these unique features, and outlines the current evidence that guides identification and treatment of patients with this condition.
38714
Online
PT60M
Hearing Loss in cCMV, in partnership with Midwestern University and Phoenix Children's Hospital
Presented by Nathan Page, MD, Aditi Bhuskute, MD
Course: #38714Level: Advanced1 Hour
AAA/0.1 Advanced; ACAud inc HAASA/1.0; AG Bell - LSLS/1.0 Domain 2; AHIP/1.0; BAA/1.0; CAA/1.0; Calif. SLPAB/1.0; IACET/0.1; IHS/1.0; Kansas, LTS-S0035/1.0; NZAS/1.0; SAC/1.0
Hearing loss in cCMV has unique features, presentation, and long-term outcomes. This presentation highlights these unique features, and outlines the current evidence that guides identification and treatment of patients with this condition.